History Of Cholesterol: Over 250 Years Of Chemistry
Jun 06, 2026
The History of Cholesterol: From Mummies and Gallstones to ApoB and Gene Editing
I have spent my career explaining cholesterol to patients, and one thing I have learned is that the whole subject makes far more sense when you understand how we got here. Cholesterol did not become controversial overnight, and the science behind it is not some recent invention dreamed up to sell statins. It took more than 250 years of chemistry, autopsies, rabbit experiments, ultracentrifuges, a long parade of Nobel Prizes, and enormous clinical trials to arrive at what we now understand about why people have heart attacks. This is the full story, from ancient mummies all the way to gene editing, because the history explains exactly why I practice the way I do today. Settle in, because this is a long one, and it deserves to be.
Ancient Cholesterol: Atherosclerosis Is Millions of Years Old
Heart disease did not arrive with fast food and seed oils. Atherosclerosis has been around for millions of years. Researchers have found evidence of atherosclerotic heart disease in mummies that lived more than 4,000 years ago. They used very high-tech imaging methods, essentially modern CT scanning, to visualize the coronary anatomy of the hearts of mummified remains, including remains from the pyramids. The studies all reached the same conclusion. Atherosclerosis was present in these mummies, and it is most likely what killed many of them.
The landmark work here is the Horus study, which examined a cohort of 137 mummies from several different ancient populations spread across the globe, from ancient Egypt to Peru to the Aleutian Islands. Probable or definite atherosclerosis showed up in roughly a third of them. These were people who ate whole foods, walked everywhere, and had none of the conveniences we now blame for modern disease, and they still developed calcified plaque in their arteries. Atherosclerosis was not picky about culture, diet, or geography. ASCVD does not discriminate. They all ate organic and hunted their food.
They also lived very short lives. The average age at death in that mummy cohort was somewhere in the range of 36 to 45 years. So when an influencer tells you that atherosclerosis is purely a disease of modern living, the archaeological record flatly disagrees. The biology of cholesterol entering an artery wall has been with us the entire time. What has changed is that we now live long enough for that slow process to actually kill us, and we finally understand the mechanism well enough to interrupt it. We actually can completely prevent heart disease now.
Infographic Summary:
Discovering Cholesterol: A Mysterious Waxy Substance
Fast forward a few million years. Long before anyone connected it to the heart, cholesterol had to be discovered as a chemical in the first place. Interestingly, as far back as 1665, Robert Boyle, the same Boyle of Boyle's law, had already described a fat transport system in animals. But the molecule itself came later.
Cholesterol was first discovered in 1769 by a French physician and chemist named Francois Poulletier de la Salle. He found it in gallstones, which are the hard deposits that form in the gallbladder. He actually isolated the crystals about a decade earlier, around 1758, by dissolving powdered gallstones in warm alcohol, but his work was never formally published, so we know about it mainly through his colleagues, including the famous chemists Macquer and Fourcroy. At this point there was no connection to heart disease at all. He simply knew he had found something new and waxy.
In 1815, a French chemist named Michel Eugene Chevreul rediscovered cholesterol and named it "cholesterine," from the Greek words chole, meaning bile, and stereos, meaning solid. He recognized that this fatty material from human gallstones could not be saponified, meaning it could not be turned into soap the way ordinary fats can, which set it apart as a distinct class of substance. Here is a fun detail I love to share: Chevreul is one of the 72 famous French scientists whose names are inscribed on the Eiffel Tower in Paris. He also lived to be 102 years old, which is its own quiet endorsement of a life spent thinking about chemistry. The modern name "cholesterol" came later still, in 1894, when the German chemist Eugen Baumann added the chemical suffix for an alcohol. So the word itself literally means solid bile alcohol.
Working Out the Structure and Where Cholesterol Comes From
Naming a molecule and understanding it are two very different things. Cholesterol turned out to be one of the most stubborn chemical structures in the history of organic chemistry. It took brilliant minds the better part of a century to figure out its architecture, and they made spectacular mistakes along the way.
Two German chemists led the charge. Heinrich Wieland received the Nobel Prize in Chemistry in 1927 for his work on bile acids and their relationship to sterols, and Adolf Windaus received the Nobel Prize in Chemistry in 1928 for his research on the constitution of the sterols and their connection with other natural substances. Here is the humbling part. The structure of cholesterol that Wieland and Windaus proposed in the late 1920s, the very work that earned them their prizes, was later shown to be wrong. The corrected structure was not published until 1932. Even Nobel laureates get the details wrong sometimes, and good science eventually fixes itself. That is a feature, not a bug.
An equally important question was where cholesterol in the body actually comes from. People assumed for a long time that the cholesterol in your blood came mostly from the cholesterol in your food. In 1933, Rudolph Schoenheimer ran a beautifully simple experiment. He placed mice in sealed containers, fed them a diet completely free of cholesterol, and then measured the cholesterol content afterward. It increased. The mice were manufacturing cholesterol out of thin air, so to speak, from other building blocks. This was an early and powerful demonstration that the body makes its own cholesterol, and that dietary cholesterol is only part of the story. To this day, this is why I tell patients that the cholesterol in your eggs is far less important than how your own liver (and every other cell in your body) handles cholesterol.
The full synthesis pathway was finally worked out by Konrad Bloch and Feodor Lynen, who shared the Nobel Prize in Physiology or Medicine in 1964 for discovering the mechanism and regulation of cholesterol and fatty acid metabolism. They showed that the body builds cholesterol through a long chain of roughly 30 enzymatic steps, starting from a tiny two-carbon building block called acetate. Understanding that pathway was the foundation for everything that followed, because if you know exactly how the body makes cholesterol, you know exactly where a drug might block it. Remember that point, because it becomes the entire basis of the statin. Your cells only need one citrate and acetate molecule to synthesize, cholesterol. And they already have both and can make their own cholesterol without any help. They just make too much.
The Rabbits That Changed Everything
The link between cholesterol and arteries started forming in the middle of the 19th century. In 1857, the German pathologist Rudolf Virchow examined diseased arteries and proposed that the lesions involved an accumulation of this same cholesterine. That was a remarkable insight for its time. But the experiments that truly nailed it down involved rabbits, and this is one of my favorite chapters in the entire story.
Between 1907 and 1909, Alexander Ignatowski, who worked in the laboratory of the Nobel Prize winner Ivan Pavlov, set out to investigate whether a diet of excess protein was toxic and accelerated aging. He fed rabbits large amounts of meat, eggs, and milk. The diet was indeed toxic for young rabbits, while adult rabbits developed atherosclerosis. Because atherosclerosis was considered a hallmark of aging at the time, Ignatowski believed his hypothesis about aging had been proven. He had stumbled onto something far bigger than he realized.
Then, in 1910, Adolf Windaus, the same chemist who would later win his Nobel Prize, reported that the plaques in aortas taken from atherosclerosis patients contained roughly 20 times more cholesterol than normal aortas. The cholesterol was concentrated right there in the disease.
The cleanest and most decisive experiment came in 1913, when Nikolaj Anitschkow, working in Saint Petersburg, discovered that a rabbit fed pure cholesterol would develop atherosclerotic cardiovascular disease, and that the disease appeared specifically when its blood cholesterol levels became greatly elevated. The degree of arterial disease tracked with how much cholesterol the animals took up. The rabbit artery turned out to be one of the best animal models we have ever had for studying human atherosclerosis. Think about what these experiments established together. The cholesterol was in the plaque. Feeding cholesterol raised blood cholesterol. And raised blood cholesterol produced the disease. That triangle of evidence was now beyond reasonable doubt, more than a century ago.
Diet Enters the Picture
As early as 1916, scientists realized that diet plays a role in cholesterol levels in humans, not just in rabbits. A scientist named Cornelis de Langen observed that people from Indonesia had much lower cholesterol levels compared to the Dutch colonists living among them. In 1922, he conducted a study to see how diet affected cholesterol. He found that when Indonesians ate a Dutch diet high in eggs and meat for three months, their cholesterol levels increased by an average of 27 percent. And Indonesians who had moved to Amsterdam ended up with the same high cholesterol levels as the Dutch themselves. The environment and the plate, not just the genes, were clearly involved. This was an early hint of something we now understand in great detail, which is that both what you inherit and how you live contribute to your cholesterol.
A Genetic Clue: Familial Hypercholesterolemia
While the diet researchers were watching cholesterol rise and fall with the menu, an entirely different line of evidence was emerging from families. In 1938, a Norwegian clinician named Carl Muller described a striking pattern. He reported families in which very high blood cholesterol, fatty deposits in the skin and tendons called xanthomas, and heart attacks in young people all traveled together. He recognized that this was an inborn error of metabolism, a condition caused by a single gene defect, and that it was passed down as a dominant trait. He had described what we now call familial hypercholesterolemia, or FH.
In 1964, a physician named Khachadurian, working with Lebanese families, took the next crucial step. He showed that FH comes in two forms. People who inherit one copy of the defective gene, the heterozygous form, have cholesterol levels about twice normal and tend to have heart attacks in adult life. People who inherit two copies, the much rarer and far more severe homozygous form, have cholesterol levels four to eight times normal and can suffer heart attacks in childhood. The heterozygous form is not rare at all. It affects roughly 1 in 500 people across most ethnic groups, and among people who have heart attacks before age 60, a meaningful fraction carry it.
I want you to sit with the importance of this. Here was a group of people whose cholesterol was extremely high from birth, who ate ordinary diets, and who developed devastating heart disease decades earlier than everyone else, in direct proportion to how high their cholesterol was. Nature had run an experiment that no ethics board would ever allow. FH would become the key that unlocked the entire mechanism, because to understand why these patients had so much cholesterol in their blood, two young scientists in Texas would have to discover exactly how the body clears cholesterol in the first place. We will get to them shortly.
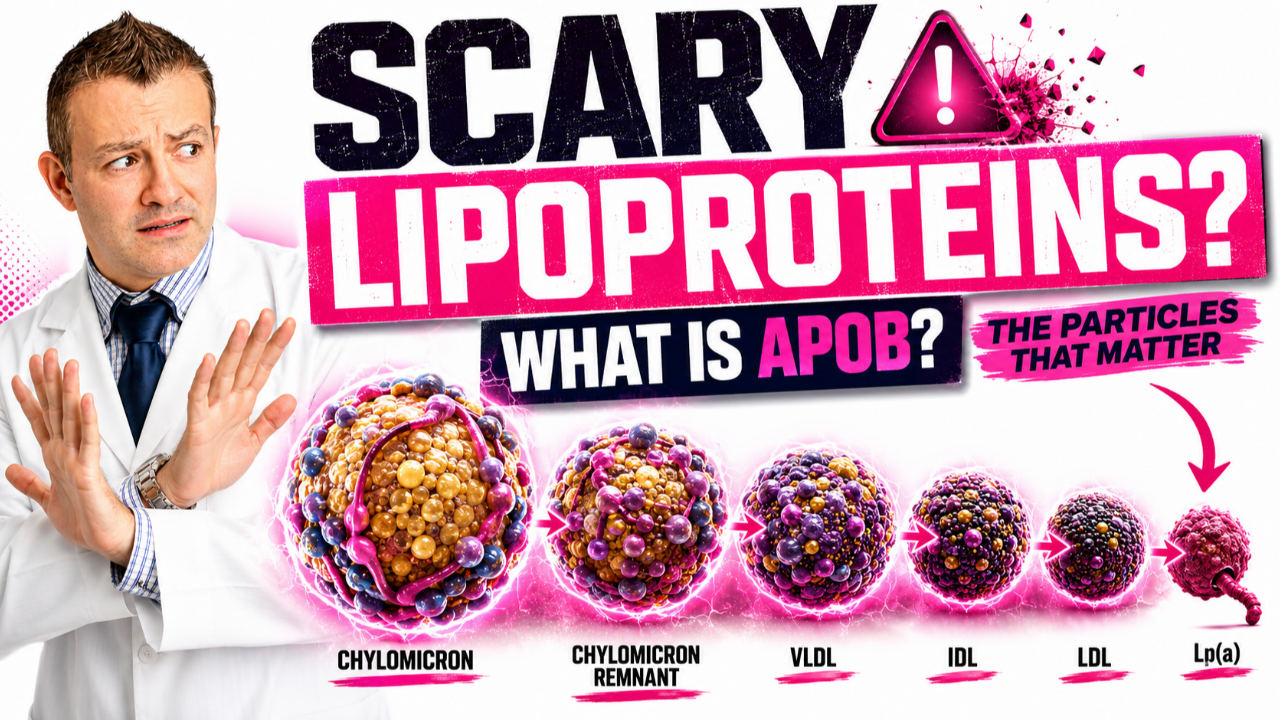
Sorting Out the Particles
By the 1920s a new puzzle was front and center. Cholesterol is an oil, and blood is mostly water, so how does it actually travel? The answers came particle by particle.
In 1924, the scientists Simon Henry Gage and Pierre Augustine Fish discovered tiny particles in human blood after a fatty meal. These particles, which they could see giving the blood a milky appearance, were about 1 micrometer in size, and we now call them chylomicrons. They are the vehicles that carry dietary fat out of the gut.
The conceptual breakthrough came in 1939, when Arne Tiselius was the first to discover that cholesterol and phospholipids do not circulate freely in plasma the way glucose does. Instead, they travel bound to proteins, specifically to the alpha and beta proteins, but not the gamma proteins. These were subsequently named alpha and beta lipoproteins. The idea that lipids ride around inside protein-wrapped vehicles, rather than floating loose, changed everything about how we think about cholesterol in the blood.
Then, in the late 1940s, John Gofman was the first to separate and further identify these lipoproteins using an ultracentrifuge. By spinning blood at enormous speeds, he could sort the particles according to their density and label them accordingly: very low density, intermediate density, low density, and high density. This is where the alphabet soup that confuses so many of my patients was born. VLDL, IDL, LDL, and HDL. I cannot stress this enough. These are not four different cholesterols. They are four different delivery vehicles, each carrying cholesterol and other fats around the body, distinguished mainly by their density and the proteins on their surface.
Lipoprotein(a): The Stubborn Outlier
There is one more particle that deserves its own paragraph, because it is having a major moment in cardiology right now and most people have never heard of it. In 1963, the Norwegian physician Kare Berg described a genetic variation in LDL that he detected using antibodies. He attributed it to a new antigen he called Lp(a), pronounced "L P little a."
Lp(a) is essentially an LDL particle, complete with its ApoB protein, that has an extra protein called apolipoprotein(a) stuck to it. Your level is set almost entirely by your genes, it stays remarkably stable throughout life, and diet and exercise barely move it. For decades it was treated as a curiosity. We now recognize, supported by both observational data and the genetic studies I will describe later, that elevated Lp(a) is an independent and causal risk factor for heart attacks, strokes, and aortic valve disease. This is why I now recommend that essentially every adult have their Lp(a) measured at least once in their lifetime. Berg discovered it more than 60 years ago, and we are only now developing drugs that specifically target it.
The 1950s and the Birth of the Risk Factor
The link between cholesterol and heart disease was strongly suspected again in the 1950s, when researchers found that people with high cholesterol levels in the blood were more likely to have heart attacks. That is when things really started to clear up for the general population, not just for families with FH.
In 1948, the United States launched the Framingham Heart Study in a single Massachusetts town, and it is still running today, now following the grandchildren of the original participants. Framingham gave us the very term "risk factor." It established that elevated cholesterol, high blood pressure, smoking, diabetes, and a sedentary lifestyle all raised the odds of cardiovascular disease. Cholesterol was formally named a cardiovascular risk factor in 1961. The entire framework that your doctor uses to estimate your risk traces directly back to this one town.
Around the same time, Ancel Keys began the work that became the Seven Countries Study, the world's first multi-country epidemiological study, comparing diet, cholesterol, and heart disease across populations on multiple continents. Keys remains a lightning rod for controversy, and the wellness internet loves to attack him, usually with the accusation that he cherry-picked data to blame saturated fat. The honest version is more nuanced. His work was imperfect, his famous equation lumped natural saturated fat together with industrial trans fat, and the dietary story turned out to be far more complicated than he framed it. He deserves the criticism on those specific points. But here is what the critics conveniently leave out. The broader body of epidemiology, from many investigators and many countries and not just from Keys, kept pointing in the same direction. Populations with higher blood cholesterol consistently had more coronary disease. You can fairly criticize Ancel Keys and still accept the mountain of independent evidence linking cholesterol to heart disease. Both things are true at once, and good critical thinking holds both.
Wellness influencers and trolls can't get over the fact that Keys discovered this in the 1950s. Set aside Keys if you disagree with him, we have 76 years of newer evidence that confirms his findings and add even more data. Let's say Key was wrong. We have over 76 years of data since then. Are we going to keep denying the evidence?
And if you go back prior to Keys, we now have over 250 years of research and science on cholesterol. Are you going to deny over 250 years of evidence?
That's up to you.
Good Cholesterol, Bad Cholesterol, and Why Those Labels Are Wrong
In the 1960s, we discovered that there are two main types of cholesterol as measured in the blood: LDL cholesterol, which got called the "bad" cholesterol, and HDL cholesterol, which they thought of as the "good" cholesterol. Obviously we do not really call them good or bad anymore, but the names have stuck around, and I still use them when first explaining cholesterol to patients because they are familiar.
Here is the truth that I want every patient and every physician to internalize. Cholesterol is just cholesterol. There is only one cholesterol molecule. It is neither good nor bad. The lipoprotein that the cholesterol is transported inside of has been described for decades as either good or bad depending on the role it happens to be carrying out at the time. And that role can change throughout the particle's lifecycle. These carriers are called lipoproteins. If lipoproteins participate in reverse cholesterol transport, they are generally considered good. Because of epidemiological data that linked LDL cholesterol to cardiovascular risk and HDL cholesterol to protection, the particles were respectively labeled bad and good.
Reverse cholesterol transport is when lipoproteins bring cholesterol back to the liver for elimination. That cholesterol can originate in various organs, including your coronary arteries. To be carried in the blood, which is mostly water, lipids like cholesterol and other fats are packaged inside protein-covered particles called lipoproteins, which again are neither inherently good nor bad.
Lipoproteins, which originate in the intestine or the liver, can deliver their lipid content to all organs in a process called forward cholesterol transport, with one important exception. They do not supply the brain, because the brain produces all of its own cholesterol behind the blood-brain barrier. The process by which lipoproteins carry cholesterol back to the liver or intestine is the reverse cholesterol transport I just mentioned.
The term reverse cholesterol transport is widely misunderstood. Initially it was thought to be the simple process by which high density lipoproteins, the HDLs, removed cholesterol out of arterial plaques and returned it to the liver for processing. We now know that the process is immensely more complicated. HDL particles can carry their cholesterol to the liver, the intestines, or to other organs such as the adrenal gland or the gonads, meaning the ovaries and testes. And we now recognize that LDL particles are just as important as HDL particles in returning cholesterol, which makes it rather silly to call LDL particles or HDL particles good or bad in any blanket way.
Where LDL particles got the name bad relates to the fact that they are the major lipoprotein that can enter the arterial wall and initiate the early steps of atherosclerosis. However, much of the cholesterol that LDL particles carry was actually acquired by transfer from HDL particles, which once again makes the good and bad labels erroneous. The labels reflect outdated beliefs that the particles returning cholesterol to the liver were good and those delivering cholesterol to the arteries were bad. We now know that not all HDL particles function properly at all times, so they are not all good, and that many, if not most, LDL particles simply return cholesterol to the liver and are, in that role, perfectly good.
How Cholesterol Actually Damages Your Arteries
Let me make the mechanism concrete, because this is where the rubber meets the road. Low density lipoproteins, the LDLs, can cross the arterial wall and, through a very complex process, deposit cholesterol inside the artery and form plaque. Lipoprotein particles usually enter the arterial wall when their concentration in the blood is high, which is most often evaluated by the blood test we call LDL cholesterol. If you have elevated LDL cholesterol, this problem is worse and it happens faster. It is fundamentally a numbers game. The more atherogenic particles you have circulating, the more of them get trapped in the wall over time.
High density lipoproteins, the HDL, can also cross the arterial wall, and they can perform good functions there by removing cholesterol and suppressing inflammation. But they can also, at times, fail to function properly, in which case these dysfunctional HDL particles could reasonably be considered bad. This is a big reason a high HDL number on your lab report is not the golden ticket people once believed it was.
When cholesterol is deposited into the arteries, it damages the arterial wall and can do one of two things. It can extend plaque into the lumen, the channel where blood flows, and gradually obstruct that flow. Or, and this is the more dangerous scenario, it can cause the plaque to erode or rupture, which triggers a clot to form, and that clot can suddenly and completely obstruct blood flow and lead to a heart attack or a stroke. Many heart attacks are not caused by the slow narrowing you might imagine, but by the sudden rupture of a plaque that was not even severely blocking the artery the day before.
Cholesterol can also be deposited in many other arteries beyond the heart, including those in your legs, your abdomen, your neck, the abdominal aorta, the renal arteries that feed your kidneys, the intestinal arteries, and the arteries of the brain, among others. For this discussion I am staying focused on the heart, but the same biology plays out throughout your entire vascular tree. High density lipoprotein helps to remove excess cholesterol from all these tissues, including plaque in the arteries. Having normal or slightly higher levels of HDL was previously thought to be straightforwardly protective against heart disease. We no longer believe this to be simply true, and the dramatic failure of drugs that raised HDL, which I will tell you about shortly, is a big part of why.
The Breakthrough: The LDL Receptor
Now we return to those families with familial hypercholesterolemia, because the mechanism behind everything came together in one of the great detective stories of modern medicine. In 1972, two young scientists at the University of Texas, Michael Brown and Joseph Goldstein, encountered two young siblings, ages six and eight, who had been hospitalized with recurrent heart attacks. These children had the severe homozygous form of FH, and their LDL levels were roughly eight times above normal. Each LDL particle was structurally normal. There were just eight times too many of them.
Brown and Goldstein asked the right question. Why could these children not clear the LDL out of their blood? Studying cells in culture, in 1973 they discovered the LDL receptor. They showed that the surface of liver cells is studded with these receptors, which grab LDL particles out of the blood and pull them into the cell in an elegant process called receptor-mediated endocytosis. The number of receptors controls how much LDL stays in circulation. Patients with FH had a deficiency of working LDL receptors, so the LDL simply piled up in their blood with nowhere to go.
Here is the part that made it world-changing. They discovered the regulation. When there is plenty of cholesterol inside a cell, the cell makes fewer receptors and clears less LDL. When the cholesterol inside the cell drops, the cell responds by making more receptors, which then clear more LDL out of the blood. This insight, that you could trick the liver into pulling more LDL out of the bloodstream by lowering the cholesterol inside its cells, is the conceptual engine behind statins and PCSK9 inhibitors alike. Brown and Goldstein were awarded the Nobel Prize in 1985 for this work, and the LDL receptor gene was cloned in the 1980s, opening the door to understanding FH at the molecular level. Suddenly we understood not just that LDL was dangerous, but precisely how the body regulates it, and crucially, how we might intervene to lower it.
A Japanese Scientist, a Mold, and the Statin Era
While Brown and Goldstein were working out the receptor, a Japanese biochemist named Akira Endo was hunting through fungi. As a young man on a farm in northern Japan, Endo had become fascinated by molds, and he had been deeply inspired by the story of how Alexander Fleming discovered penicillin in a mold. He became convinced that molds might hold other medical treasures.
Working at the Sankyo laboratories, Endo set out to find a compound from a fungus that would block cholesterol synthesis, targeting that exact pathway Bloch and Lynen had mapped. He screened more than 6,000 fungal strains. In 1973, from a blue-green mold, he isolated compactin, the first compound that inhibited HMG-CoA reductase, the rate-limiting enzyme of cholesterol synthesis. This is the foundation of every statin ever made.
The road from discovery to medicine was anything but smooth, and the story is full of moments where statins almost died in the lab. When Sankyo first tested compactin by feeding it to rats, it did not lower cholesterol at all, and the project nearly stalled. Endo did not give up. He later tried it in laying hens, then in dogs and monkeys, and in those animals it produced dramatic reductions in cholesterol. Sankyo's executives showed little interest in human studies, so Endo set up his own collaboration with a physician named Akira Yamamoto, who in 1978 gave compactin to a small group of patients with severe familial hypercholesterolemia and saw their cholesterol fall by about 30 percent, the largest reductions anyone had ever seen in those patients.
Then came the scare that nearly killed the entire class of drugs. In 1980, clinical development of compactin was halted because the drug was suspected of causing lymphomas in dogs that had been given enormous doses, on the order of 100 to 200 times a human dose, for two years. Because the new Merck compound was so similar, this scare temporarily froze the development of the first statin to reach market, too. Merck, which had obtained samples of compactin from Endo and then discovered its own remarkably similar molecule from a different mold, a molecule that differed from compactin by only a handful of atoms, paused its program in alarm. Experts in the field urged caution but also urged Merck not to abandon the work, pointing out that the doses involved were absurdly high and that the Japanese FH patients were experiencing life-changing benefits.
Merck pressed forward with careful new long-term studies. By the mid-1980s, those studies confirmed that the drug, now called lovastatin, dramatically reduced cholesterol, was well tolerated, and produced no tumors at sensible doses. In September 1987, lovastatin became the first statin approved by the FDA. Other cholesterol drugs had existed before it, such as the bile acid binder cholestyramine, which was a gritty powder that tasted terrible, and the fibrate gemfibrozil, which the Helsinki Heart Study showed in 1987 reduced heart attacks but never proved a survival benefit. Statins were about to leave all of them behind.
Then came the trial that ended the debate once and for all. In 1994, the Scandinavian Simvastatin Survival Study, known as 4S, enrolled 4,444 patients with established heart disease. Simvastatin did not merely lower cholesterol numbers on a lab report. It reduced heart attacks, and it reduced death. The trial was actually stopped early because the survival benefit was so clear and it would have been unethical to keep some patients on placebo. That was the moment statins went from promising to essential.
Akira Endo is rightly called the father of statins, and a later member of the class, atorvastatin, went on to become the best-selling drug in the history of medicine. Endo himself, fittingly, passed away in 2024 at the age of 90, having lived to see the medicines born from his moldy cultures save many millions of lives.
When the "Good Cholesterol" Drugs Failed
I promised you the story of why we stopped worshipping HDL, and it is one of the most instructive failures in all of cardiology. If high HDL was protective, the logic went, then a drug that raised HDL should prevent heart attacks. Drug companies poured enormous resources into a class of medications called CETP inhibitors, designed to raise HDL substantially.
The first of them, torcetrapib, raised HDL impressively. It also increased deaths and cardiovascular events in its large trial, which was halted. Later CETP inhibitors avoided that specific toxicity but largely failed to deliver the expected benefit, with one trial after another stopped for futility. Raising the HDL number simply did not rescue patients the way the theory predicted. The "good cholesterol" hypothesis, at least in its simple form, collapsed under the weight of the evidence.
This is the part I want you to appreciate as a thinker, not just as a patient. The collapse of the HDL hypothesis was confirmed by a completely independent line of evidence. Researchers found people who carry genes that give them naturally high HDL their whole lives, and they discovered that those people did not have lower rates of heart attacks. If lifelong high HDL were truly protective, those individuals should have been protected, and they were not. The conclusion was unavoidable. It is not the HDL number that matters. What matters is the burden of atherogenic, ApoB-containing particles, the LDL and its cousins, that get into the artery wall. This realization is exactly why I focus on ApoB rather than chasing an HDL number, and it is a direct lesson from one of the most expensive failures in pharmaceutical history.
The Genetic Proof: Nature's Own Randomized Trial
For a long time, skeptics could argue that cholesterol was merely associated with heart disease without truly causing it. That argument is now dead, and it was killed by genetics. The technique is called Mendelian randomization, and it is one of the most elegant tools in modern medicine.
Here is the idea. At conception, we each inherit a random assortment of gene variants, including variants that set our LDL slightly higher or slightly lower for life. Because this assortment is essentially random, nature has effectively run a giant randomized trial, sorting millions of people into lifelong-lower-LDL and lifelong-higher-LDL groups from birth. When researchers compare these groups, the finding is overwhelming and consistent. People who inherit lifelong lower LDL have dramatically less heart disease, and the benefit is far larger per unit of LDL than what we see in short drug trials. The same approach validated the danger of Lp(a) and confirmed that the LDL receptor pathway, the PCSK9 pathway, and others all converge on the same conclusion. LDL and the ApoB particles that carry it are causal.
This led to one of the most important concepts in preventive cardiology, championed by researchers like Brian Ference and the legendary Eugene Braunwald: cumulative LDL exposure, sometimes described as the area under the curve. Your risk is not just about your LDL today. It is about your LDL multiplied by all the years you have carried it. A person with moderately high LDL for 50 years can accumulate the same arterial burden as a person with very high LDL for a shorter time. This is why the FH patients have heart attacks so young, and it is why I am such a strong advocate for getting LDL and ApoB under control early in life rather than waiting until your 60s. The damage is cumulative, and time is the multiplier you cannot get back.
Beyond Statins and Into the Modern Era
The story did not stop with statins, and this is where my excitement really takes over, because the pace of progress in the last two decades has been breathtaking. We added ezetimibe, which blocks cholesterol absorption in the gut and lowers LDL through a completely different mechanism, making it a natural partner to statins.
Then came one of my favorite chapters of all. In 2003, researchers led by Abifadel and colleagues, studying families with extreme cholesterol levels, identified a gene called PCSK9. This protein is essentially the saboteur of the LDL receptor. PCSK9 marks LDL receptors for destruction, so the more PCSK9 you have, the fewer receptors survive and the more LDL stays in your blood. Some families had overactive PCSK9 and sky-high cholesterol. Remarkably, other people were found to carry a broken PCSK9 gene, which gave them very low LDL their entire lives and strong protection from heart disease, with no apparent downside. That natural experiment told scientists exactly what to do: block PCSK9 and let the liver keep more of its receptors.
In 2015, the first PCSK9 inhibitors, evolocumab and alirocumab, were approved. These are injectable antibodies, and large outcome trials such as FOURIER confirmed that they cut cardiovascular events on top of statins while driving LDL to levels we once assumed were impossible to reach safely, sometimes down around 30 milligrams per deciliter, with no lower threshold of harm appearing.
Today we are watching the next leap arrive in real time. We now have inclisiran, a small interfering RNA therapy that silences the PCSK9 gene with only two injections per year. We have bempedoic acid for patients who cannot tolerate statins. And we have early human gene editing trials, including programs aiming to switch off PCSK9 permanently with a single treatment that could lower LDL for a lifetime. Every one of these advances rests on the same chain of insights I have walked you through: Bloch and Lynen mapped the synthesis pathway, Brown and Goldstein found the receptor and its regulation, Endo found the molecule that exploits it, and the geneticists proved the whole thing is causal. It is the cumulative burden of ApoB-containing particles, accumulated over a lifetime, that drives atherosclerosis. The lower and the earlier we keep that burden, the less heart disease we see.
Understanding Your Lipid Panel
After 250 years of science, here is what actually lands on your lab report, and how to read it. Cholesterol is not a one-size-fits-all substance. There are multiple different parts and multiple different particles and types, and this is exactly why your panel can look so confusing. When you get your standard lipid panel, you will typically see it divided up as follows:
Total Cholesterol (TC)
LDL Cholesterol (LDL-C)
HDL Cholesterol (HDL-C)
Triglycerides (TG)
Sometimes you will also get:
VLDL Cholesterol (VLDL-C)
IDL Cholesterol (IDL-C)
Non-HDL Cholesterol (non-HDL-C)
Various potential ratios, which can vary from lab to lab
Each of these has its own importance, and I dig into all of them with my patients and in my community. Notice that I am very deliberately using LDL-C to identify LDL cholesterol. You have to be specific when you use this terminology, because there are also many other lab tests that evaluate LDL lipoproteins in different ways, including LDL particle number, LDL size, LDL subfractions, LDL oxidation, and more.
The amount of cholesterol inside the particles, which is what LDL-C measures, is not the same thing as the number of particles, which is what ApoB measures. Every atherogenic particle, whether it is an LDL, a VLDL, an IDL, or an Lp(a), carries exactly one ApoB protein, so ApoB counts your total atherogenic particle burden in a single number. That distinction, particle count versus cholesterol content, is the single most important upgrade in how we think about risk today, and it is the natural endpoint of this entire 250-year story. When the two disagree, and they often do, especially in people with diabetes, obesity, or high triglycerides, the particle count wins.
Why This History Matters for You
I tell you all of this because cholesterol science is not a recent fad, and it is certainly not a marketing scheme cooked up to sell pills. It is the product of centuries of careful work, a long list of Nobel Prizes spanning chemistry and medicine, and clinical trials involving hundreds of thousands of patients. When a wellness influencer tells you that cholesterol does not matter and the whole thing is a myth, they are not revealing some hidden truth that the medical establishment is too corrupt to admit. They are ignoring 250 years of remarkably consistent evidence that has saved millions of lives.
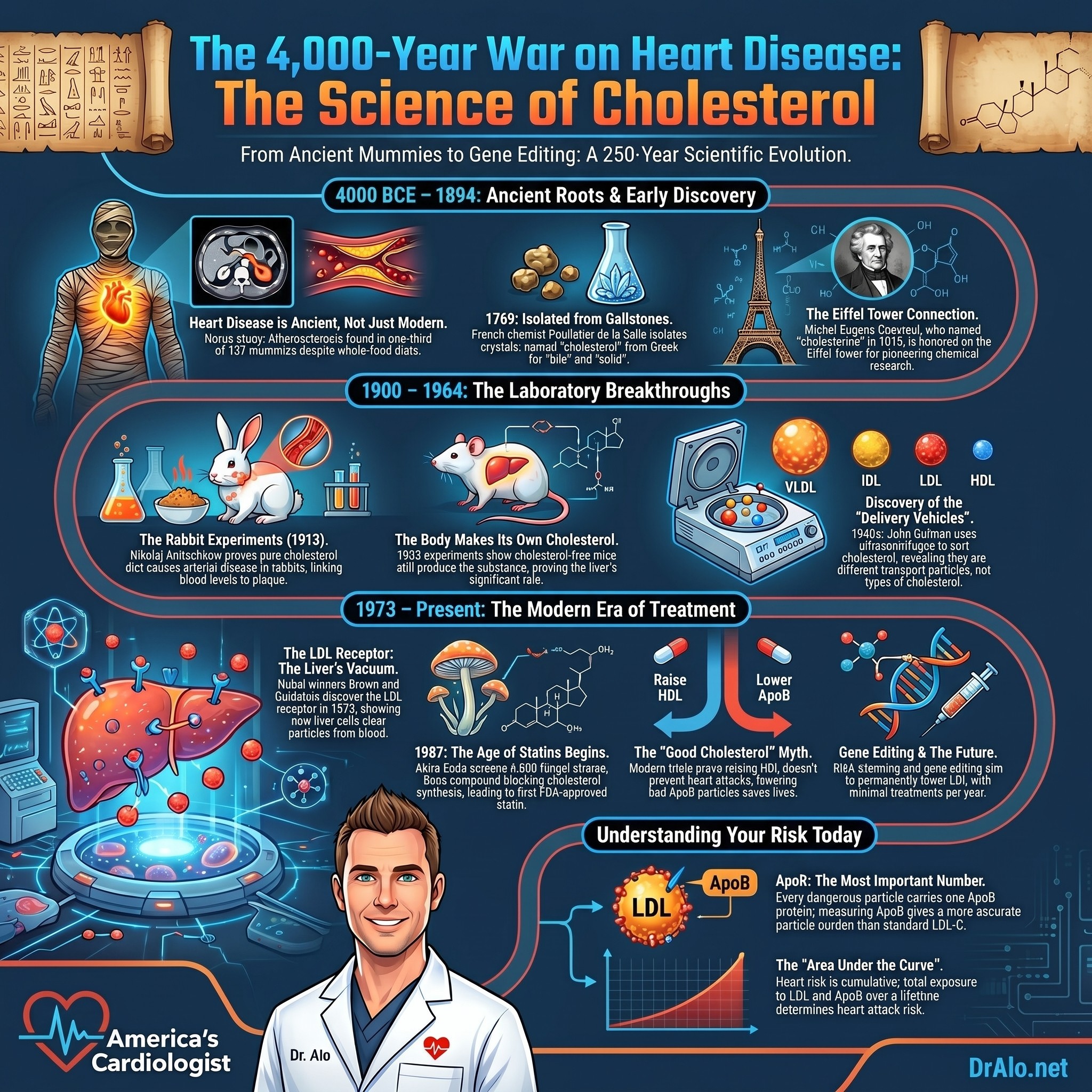
Look at the full arc. The mummies had atherosclerosis 4,000 years ago. Poulletier and Chevreul found and named the molecule, earning Chevreul a place on the Eiffel Tower. Wieland, Windaus, Bloch, and Lynen worked out its structure and how the body builds it. The rabbits of Ignatowski and Anitschkow proved the mechanism. Muller and Khachadurian showed us the genetic experiments nature had already run in human families. Tiselius and Gofman discovered how cholesterol travels. Framingham gave us the risk factor, Berg found Lp(a), and Brown, Goldstein, and Endo gave us the receptor and the medicine. The trials proved, again and again, that lowering the burden changes the outcome, and the geneticists proved that it was causal all along. That is not controversy. That is one of the great success stories in the entire history of medicine, told across centuries by hundreds of scientists who mostly never met each other, all arriving at the same answer.
If you want to understand your own numbers, especially your ApoB and your Lp(a), and learn how to actually protect your heart with evidence rather than supplements and slogans, I would love to have you in my community. Come join the conversation at https://dralo.net/community, and if you want to go deeper, register for my next free webinar at https://dralo.net/webinar.
References
Imaging of atherosclerosis in ancient human populations. Atherosclerosis literature.
Francois Poulletier de la Salle and the first isolation of cholesterol crystals from gallstones.
National Lipid Association. The Lipid Hypothesis: a historical overview.
The Seven Countries Study, directed by Ancel Keys.
The Framingham Heart Study and the origin of the cardiovascular risk factor concept. Circulation.
Lipoprotein(a): structure, function, and the discovery by Kare Berg in the 1960s.
The Nobel Prize in Physiology or Medicine 1985: Brown and Goldstein and the LDL receptor.
Lasker Foundation. Akira Endo, Brown and Goldstein, and the road to statins.
Akira Endo: Father of Statins. National Library of Medicine.
The discovery of statins: compactin, lovastatin, and the dog toxicity scare. Cell, 2008.
Akira Endo, who discovered a penicillin for heart attacks (1933 to 2024). PNAS.
Scandinavian Simvastatin Survival Study (4S). The Lancet, 1994.
A translational medicine perspective on the failure of torcetrapib and the HDL hypothesis.
Still Have Questions? Stop Googling and Ask Dr. Alo.
You’ve read the science, but applying it to your own life can be confusing. I created the Dr. Alo VIP Private Community to be a sanctuary away from social media noise.
Inside, you get:
-
Direct Access: I answer member questions personally 24/7/365.
-
Weekly Live Streams: Deep dives into your specific health challenges.
-
Vetted Science: No fads, just evidence-based cardiology and weight loss.
Don't leave your heart health to chance. Get the guidance you deserve. All this for less than 0.01% the cost of health insurance! You can cancel at anytime!
[👉 Join the Dr. Alo VIP Community Today]
Cardiology & Obesity Medicine